

こんにちは、すきとほる疫学徒です。
【初心者のためのRで医療ビックデータ解析】シリーズは、「これまでデータの解析を行ったことがない人」を対象にして、Rで医療ビックデータを解析する方法をわかりやすく解説していきます。
全てのシリーズを受講し終えた時点で、「診療報酬データやレジストリーといった医療大規模データを、自立して解析できる」状態になることを目指しております。
全研究者、Rユーザー化計画、ここに始動す。
こちらの記事はパート6になりますので、まずそれ以前のパートをお読み頂いてから、この先に進んで頂けますと、より一層Rの勉強が楽しめるかと思われます。
これまでのCodeを走らせる
さて、本日はいよいよ分析をしていきましょう。
これまで作成したデータセット、変数を使って、T検定、カイ二乗検定などの検定をしていきます。
まず、以下のCodeを走らせて、これから分析で使うデータセットを作成してください。
ただCodeを走らせるのではなく、「このCode、何をしようとしてるんだっけ?」と思い出しながら走らせていくと、復習にもなって良いですね。
library(tidyverse)
library(lubridate)
library(haven)
library(summarytools)
library(ggplot2)
library(tableone)
library(dplyr)
library(naniar)
library(devtools)
library(reader)
library(missForest)
set.seed(777)
hosp_id <- sample(1:10, size = 1000, replace = T)
sex <- sample(0:1, size = 1000, replace = T)
BMI <- round(rnorm(1000, mean = 18, sd = 2), 1)
length_of_stay <- round(rnorm(1000, mean = 30,sd = 10))
ADL <- sample(0:100, size = 1000,replace = T)
smoking_status <- sample(0:100, size = 1000,replace = T)
age <- round(rnorm(1000, mean = 60,sd = 10))
dementia <- sample(0:1, size = 1000,replace = T)
myocardial_infarction <- sample(0:1, size = 1000,replace = T)
diabetes_mellitus <- sample(0:1, size = 1000,replace = T)
liver_disease <- sample(0:1, size = 1000,replace = T)
solid_tumor <- sample(0:1, size = 1000,replace = T)
hospital_bed_size <- sample(20:500,size = 1000, replace = T)
academic_hospital <- sample(0:1, size = 1000, replace = T)
drug_A <- sample(0:1, size = 1000, replace = T)
drug_B <- sample(0:1, size = 1000, replace = T)
death <- sample(0:1, size = 1000, replace = T)
ope <- sample(0:1, size = 1000, replace = T)
year <- sample(2010:2020, size = 1000, replace = T)
df <- data.frame(hosp_id, sex, age, BMI, ADL, smoking_status, dementia, myocardial_infarction, diabetes_mellitus,
liver_disease, solid_tumor, ope, year, drug_A, drug_B, hospital_bed_size, academic_hospital,
length_of_stay, death)
df <- prodNA(df, noNA = 0.2)
# Variable creation -------------------------------------------------------
df <- df %>%
mutate(age_c = case_when(age <= 19 ~ "-19",
age > 19 & age <= 64 ~ "20-64",
age > 64 ~ "65-"))
df <- df %>%
mutate(smoking_c = case_when(smoking_status == 0 ~ "No smoker",
smoking_status >= 1 & smoking_status <= 50 ~ "Moderate smoker",
smoking_status > 50 ~ "Severe smoker"))
df <- df %>%
mutate(adl_c = case_when(ADL <= 10 ~ "Low ADL",
ADL >= 11 & ADL <= 90 ~ "Mid ADL",
ADL <= 100 ~ "High ADL"))
df <- df %>%
mutate(female = case_when(sex == "1" ~ "Yes",
sex == "0" ~ "No"))
df <- df %>%
mutate(across(c(academic_hospital,
death,
dementia,
ope,
drug_A,
drug_B,
liver_disease,
myocardial_infarction,
diabetes_mellitus,
solid_tumor),
~ case_when(. == "1" ~ "Yes",
. == "0" ~ "No"),
.names = "{.col}2_c"))
df <- df %>%
group_by(hosp_id, year) %>%
mutate(ope_count = sum(ope, na.rm = T)) %>%
ungroup()
さて、そうしましたらここで一旦、データを記述して、新たに作成したデータセットの特徴を掴んでいきましょう。
今回は、「DrugAを投与された患者と、されていない患者間で、治療効果にどのような差があるか」ということを明らかにするために、研究を行っているものとします。
ですので、論文にはおそらくTable1として、DrugAの有無で2群に分け、患者特性が記述されていくはずです。
では、このTable1を作ってみてください。
どのように作成するか、覚えてらっしゃいますか?
忘れてしまった方は、こちらから振り返ってみましょう。
CreateTableOne()を使うんでしたね。
listvars <- c("age_c", "female", "BMI", "adl_c", "smoking_c", "year",
"drug_B2_c", "ope2_c", "academic_hospital2_c",
"dementia2_c", "liver_disease2_c", "myocardial_infarction2_c",
"solid_tumor2_c", "death2_c", "length_of_stay")
listcat <- c("age_c", "female", "adl_c", "smoking_c", "drug_B2_c",
"ope2_c", "academic_hospital2_c", "dementia2_c",
"liver_disease2_c", "myocardial_infarction2_c",
"solid_tumor2_c", "death2_c")
Table1 <- CreateTableOne(vars = listvars,
factorVars = listcat,
includeNA = T,
addOverall = T,
data = df,
strata = "drug_A2_c")
Table1
こうです。
では引き続き問題。
こうして作成されたTable1は、もちろんそのままでは論文に掲載することができません。
ですので、Table1をエクセルに書き出してやる必要があります。
さて、どうするんでしたっけ?
print(Table1) %>%
write.csv("/Users/***/Desktop/R_file/Blog/Rblog/Output/Table1.csv")
こうですね。
print()で、Table1をエクセルに書き出す準備をし、そしてwrite.csv()で書き出します。
この時、
「このフォルダに書き出してくださいね」という書き出し先の住所と、
「このファイル名で書き出してくださいね」という宛名を指定してあげる必要があります。
書き出し先のフォルダですが、事前に皆さんが使っているRblogというフォルダの中に、さらにOutputというフォルダを作成しましょう。
こちらのR Tipsの8番目でも紹介しておりますが、このようにR関連のフォルダを作る場合には、Code、Backup、Data Source、Outputと種類別にフォルダを分けて保存すると良いです。
さて、フォルダができたら、その住所であるパスをコピーしましょう。
パスのコピーの仕方は、こちらの記事で書いているので、わからない方はご参照ください。
できたら、write.csv()の中に貼り付けますが、そのまえに””を忘れずに入力してください。
print(Table1) %>%
write.csv("/Users/***/Desktop/R_file/Blog/Rblog/Output")
こんな感じにいなるはず。
そしたら最後に、
/ファイル名.csv
をつけて、コードを実行してあげれば、指定したフォルダにcsvが入っているはずです。
私もいまだにやってしまうのですが、ファイル名の最後に.csvをつけるのをお忘れなきよう。
ちなみに、データセットを書き出したい場合もwrite.csv()を使います。
まず、書き出し先としてSourceフォルダを作ってやりましょう。
このセミナーではCodeからデータセットを作成しているため、外部からデータセットを読み込むという作業が発生しませんが、通常は何らかの方法で入手したデータセットをRで読み込み、解析していくことになります。
Sourceフォルダができたら、パスをコピーします。
そしたら、そのパスをこんな感じでwrite.csv()に入れ込んでやりましょう。
write.csv(df,
row.names = F,
"/Users/***/Desktop/R_file/Blog/Rblog/Source/df.csv")
上から順に、
df:書き出すデータフレームを指定
row.names = F:先頭行列に番号を挿入しない
という指示を出しています。
パス名は、
“”で囲むことと、
最後にファイル名.csvをつけることを忘れないようにしましょう。
そうすると、先ほど作成したSourceフォルダにdf.csvという名のcsvファイルが作成されているはずです。
作成したファイルは、こちらの記事で紹介しているread_csv()で読み込めますので、こうすれば外部のデータを解析にしようできるようになります。
データの記述は本記事のメインコンテンツではないので、ここまでにしますが、実際に解析をするときには、さらに欠損の状況を把握したり、グラフを描いて視覚的にデータを把握する必要があります。
それらも全て過去の記事で紹介していますので、併せてご覧ください。
2変数間の検定を行う
さて、本題である検定に辿り着くまでにちょっと時間がかかってしまいましたが、いよいよここからは検定の説明です。
T検定
まずは、アウトカムが連続値であるときに使用するT検定です。
Codeはこう。
t.test(length_of_stay ~ drug_A2_c,
data = df)
すると、このようにConsoleに結果が表示されます。
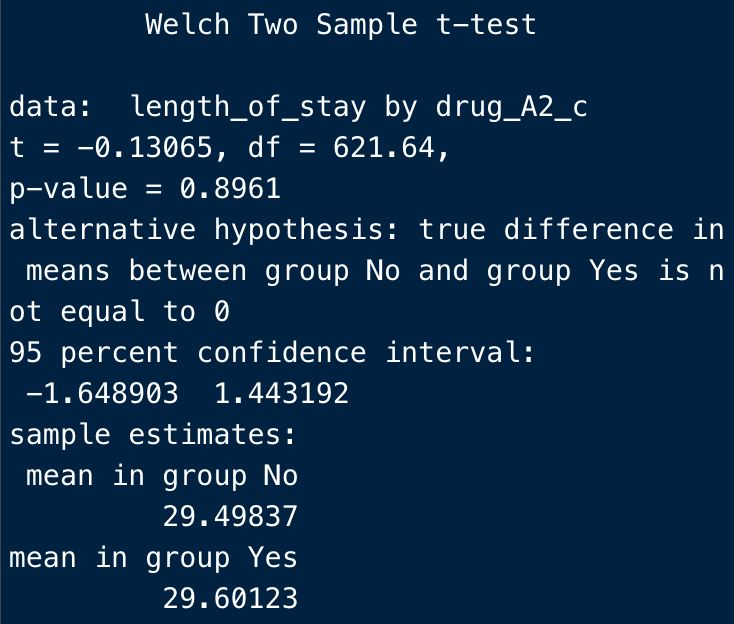
まず冒頭で、”Welch Two Sample t-test”を実施したことを示します。
今回は、データが等分散でないと仮定し、ウェルチ検定を行っています。
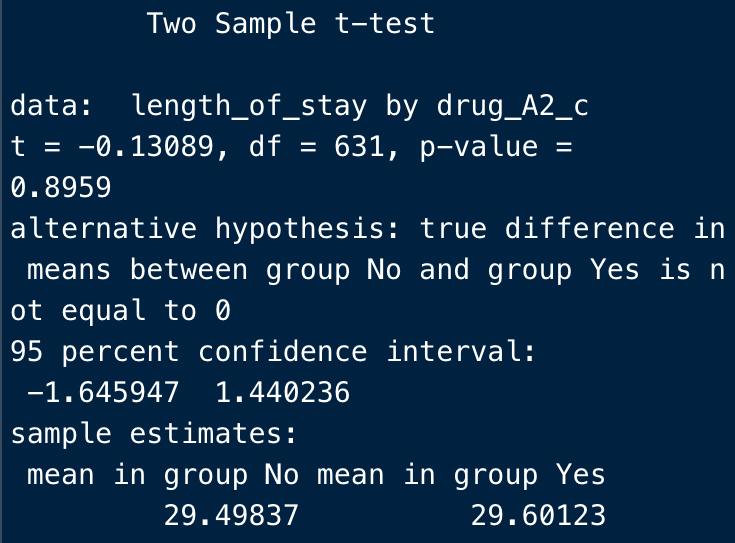
もし等分散を仮定したい場合には、このように引数にvar.equal = Tをとってやりましょう。
t.test(length_of_stay ~ drug_A2_c,
var.equal = T,
data = df)
すると、このように冒頭から”Welch”という言葉が消えているのがわかります。
結果の解釈ですが、
t:T値
P-value:P値
95 percent confidence interval:平均の差の95%信頼区間
mean:両群の平均値
を表しています。
この場合、「両群の平均値に差があるとは言えない」という結果ですね。
t.test()の引数としては、
paired = T:対応のあるT検定(デフォルトはFalse)
conf.level = 0.95:で任意の信頼区間を設定
です。
Wilcoxonの順位和検定
同じくデータが連続値の時に用いる検定ですが、Wilcoxonの順位和検定は正規分布を仮定しません。
Codeはこう。
wilcox.test(length_of_stay ~ drug_A2_c,
data = df)
ほいで結果がこう。
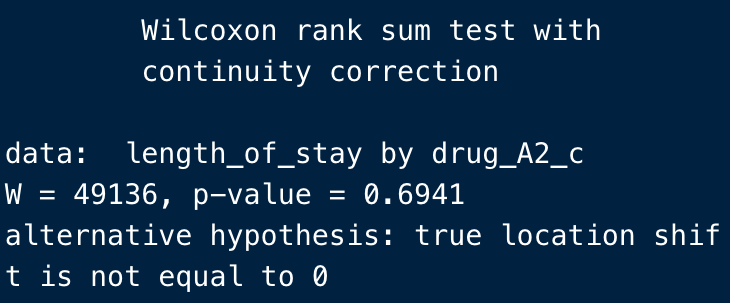
先ほどのT検定の結果よりはややシンプルですね。
こちらも、
p-valueがP値を表します。
引数はt.test()と同様で、
paired = T:対応のある検定(デフォルトはFalse)
conf.level = 0.95:で任意の信頼区間を設定
カイ二乗検定
お次はカテゴリー変数同士の検定である、カイ二乗検定。
Codeはこう。
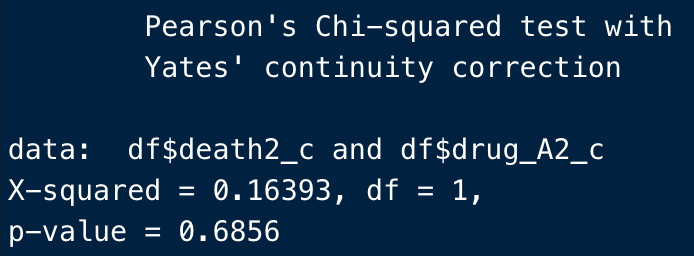
chisq.test(df$death2_c, df$drug_A2_c)
ほんで結果がこう。
Fisherの正確検定
次は、サンプルサイズが少ない時に用いるFisherの正確検定です。
Codeはこう。
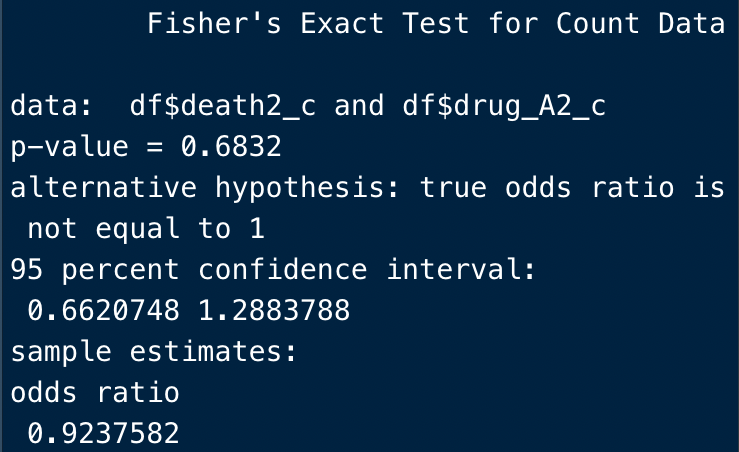
fisher.test(df$death2_c, df$drug_A2_c)
結果はこう。
2変数間の検定の補足
さて、これで2変数間の検定の基本的な方法をお伝えできました。
えー、せっかく学んで頂いたのに大変申し訳ないのですが、実はこれらの関数、解析ではほぼ使用しません。。。
既にCreateTableOne()をしっかり勉強してくださっている方はお気づきかもしれませんが、ここでお伝えした検定は、CreateTableOne()を使えば、一括で行うことができます。
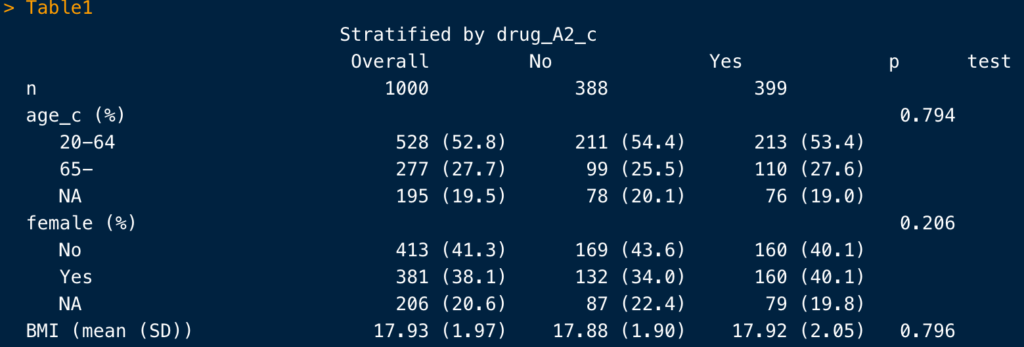
なので、Table1に検定結果を掲載する必要がある際には、CreateTableOne()で作成されたTableをそのまま掲載すれば何の問題もありません。
こちらは、CreateTableOne()で作成したTableですが、このように右端に検定結果がついています。
相関係数を求める
さて、次は2変数間の相関係数の求め方をお伝えします。
全ての変数を使うと、画面が非常にややこしいことになるので、一部の変数に絞ったデータフレームを新たに作ります。
df_n <- data.frame(lapply(df, function(x) as.numeric(x))) %>%
select(age, ADL, BMI, length_of_stay, hospital_bed_size, ope_count)
lapplyはapply一族の関数で、色々と便利なやつなのですが、今回の本題ではないので触れません。
そうしたら、cor()で相関係数を算出します。
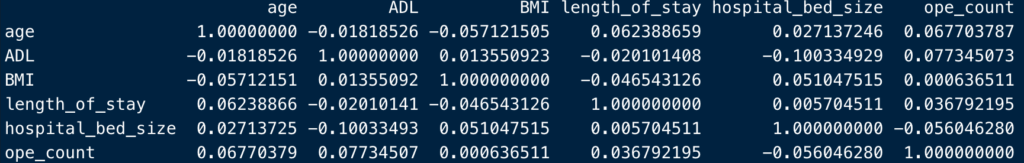
cor(df_n,
use = "complete.obs")
結果はこんな感じ。
小数点が長くて鬱陶しいので、丸めてあげましょう。
round()は、小数点何桁までを表示するかを指定します。
cor(df_n,
use = "complete.obs") %>%
round(2)
丸まりました。
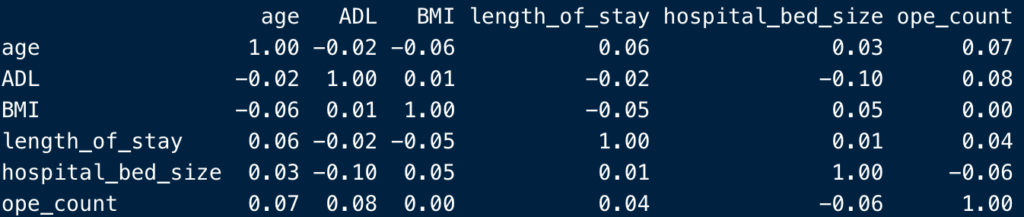
Spearmanの相関係数を求めたい場合は、このように、method = “spearman”を指定します。
cor(df_n,
use = "complete.obs",
method = "spearman")
しかしまぁ、見にくいことこの上ないですね、この結果。
もう少し直感的に相関関係を捉えられたら良いのですが。。。
安心してください、ちゃんとご希望にお答えするパッケージがあります。
とりあえずこちらのCodeを走らせましょう。
install.packages("GGally")
library(GGally)
df2 <- df %>%
select(age, female, ADL, BMI, length_of_stay, drug_A2_c)
そしたらこう!
ggpairs(df2)
すると、こんな図が出力されます。
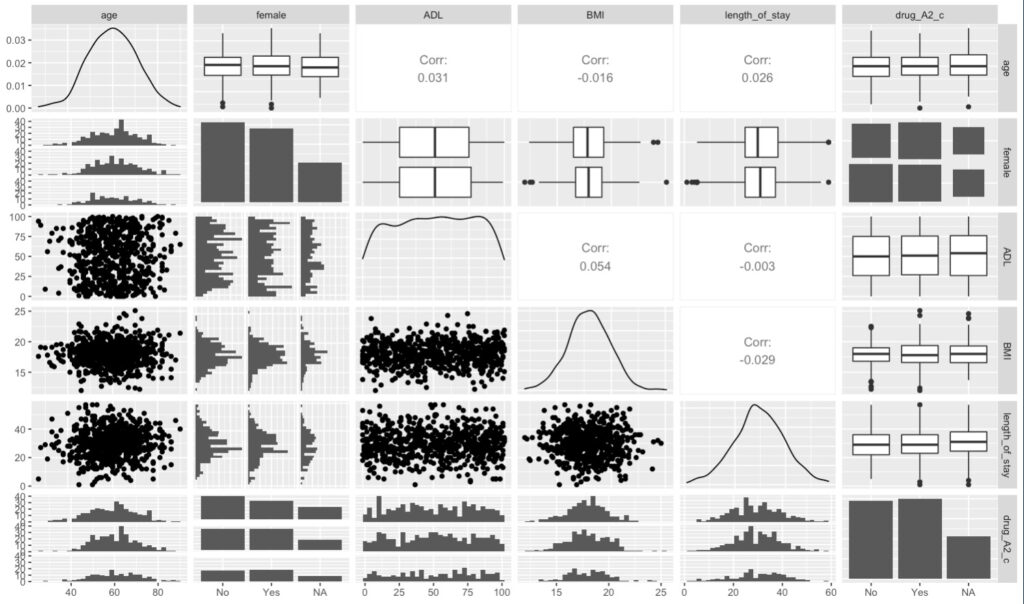
連続値 x 連続値のマスには相関係数が表示されています。
斜めを走るラインもしくは棒グラフは、各変数の密度関数です。
このように、ggpairs()は解析に打ち込んだ変数のそれぞれの関係性を、相関係数を含めて一括でVisualizeしてくれるんですね。
ほんと便利ですよねぇ。。。
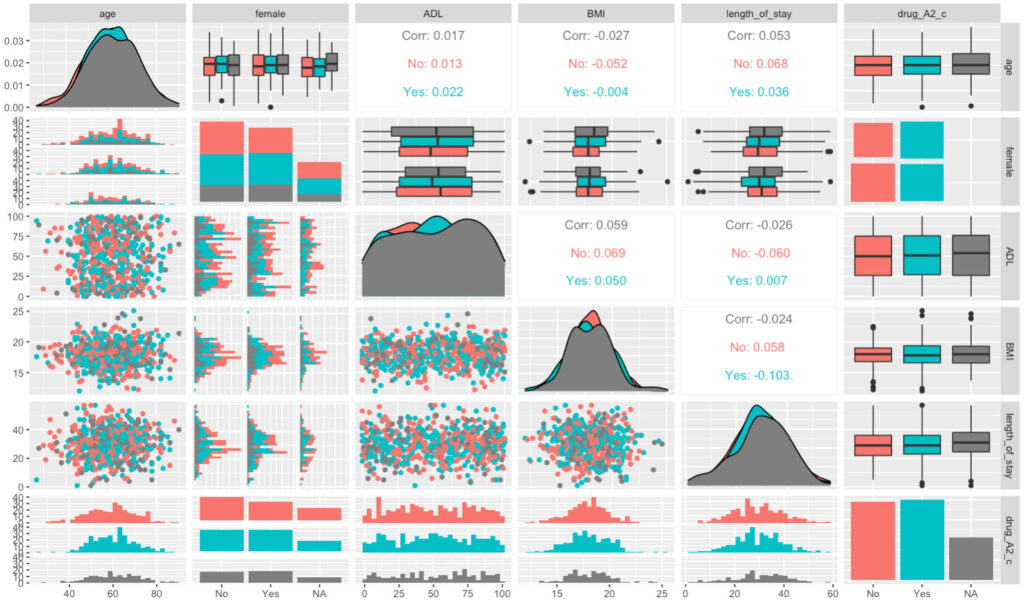
さらに、DrugAの内服の有無で層別化したい場合には、こう。
ggpairs(df2,
aes(colour = drug_A2_c))
すると、こう。
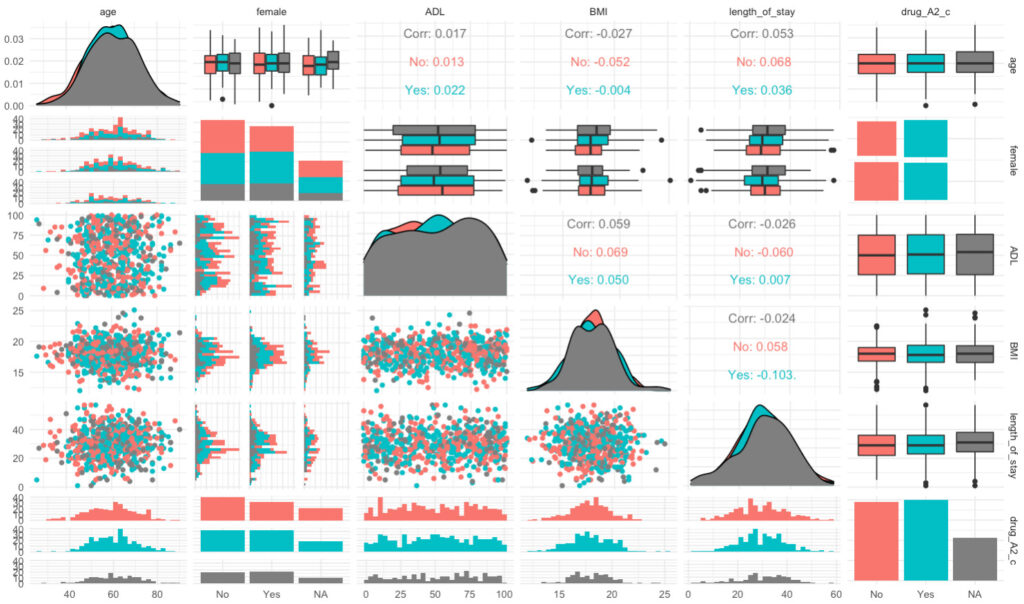
テーマを変えたければ、theme_***()を追加してあげる(***は色々あるから、お好みを探してみて)。
ggpairs(df2,
aes(colour = drug_A2_c)) +
theme_minimal()
すると、良い感じに。
他にも色々といじり方があって、詳細はこちらの記事が詳しいので、覗いてみると良いと思います。
https://assabu.exblog.jp/28933942/
終わりに
お疲れ様でした、今日はここまでです!
2群の検定だけでなく、もちろんRではそれ以上の群間の検定も行うことができます。
考え方は本日紹介した関数と大きく違いませんので、皆さんそれぞれで3群の検定を行うための関数を調べてみてください。
日本語でも英語でも、きっとすぐ参考になるページが出てくると思います。
さて、検定が終わりましたので、次はいよいよ解析に入っていきます。
単回帰、
重回帰、
多変量回帰、
生存時間分析などですね。
長かった”初心者のためのRで医療ビックデータ解析”もようやく終わりが見えてきました。
私の想定ですと、あと3回ほどで終了になると思いますので、皆様もうしばしお付き合いくださいませ。
すきとほる疫学徒からのお願い
本ブログは、全ての記事をフリーで公開しており、「対価を払ってやってもいいよ」と思ってくださった方のみに、その方が相応しいと思っただけの対価をお支払い頂けるPay What You Want方式を採用しています。
教育に投資できる方だけがさらに知識を身につけ、そうでない方との格差が開いていくという状況は、容認されるべきではないと考えているからです(そもそも私程度の記事によって知識の格差が広がると考えていることが、勘違いかもしれませんが)。
私自身も高校卒業後は大学・大学院の学費、生活費と自分で工面する中で、親の支援を得られる友人たちがバイトをせずに学習に集中したり、海外留学や旅行などの経験を積んだりする様子を見て、非常に悔しい想いをしたことを思い出します。
読者の皆様におかれましては、「勉強になった!」、「次も読みたい!」と本ブログに価値を感じてくださった場合のみ、ご本人の状況が許す限りにおいて、以下のボタンからご自身が感じた価値に見合うだけの寄付を頂戴できますと幸いです。
もちろん価値を感じなかった方、また学生さんなど金銭的に厳しい状況にある方からのご寄付は一切不要です。



